

Burrow-wheeler Transform 알고리즘은 Block-Sorting compression으로 불린다.
BWT 알고리즘은 압축을 시킬 파일의 구성 원소를 빈도 수 별로 분석 한 후 분석된 값을 효율 적으로 표현 시키는 data compression에 사용되는 알고리즘이다.
문자열이 BWT에 의해 변환되면 character 의 값은 변하지 않고 순서들만 변하게 된다.
원본 문자열에서 몇몇 substring이 자주 발생하게 되면, 변환된 문자열에서는 single character가 여러 번 반복되는 위치가 생기게 된다.
이것은 move-to-front transform과 run-length encoding에 의해 압축하기에 편리하다.

output이 input에 비해서 압축 하기가 쉬운 이유는 많은 캐릭터들 반복 해서 나타나기 때문이다.
변환이 된 output string을 보자면, 총 6개의 구분하기 쉬운 character들이 있다.
XX, SS, PP II or III이 character들은 총 44개의 character 중에 13개를 차지한다.
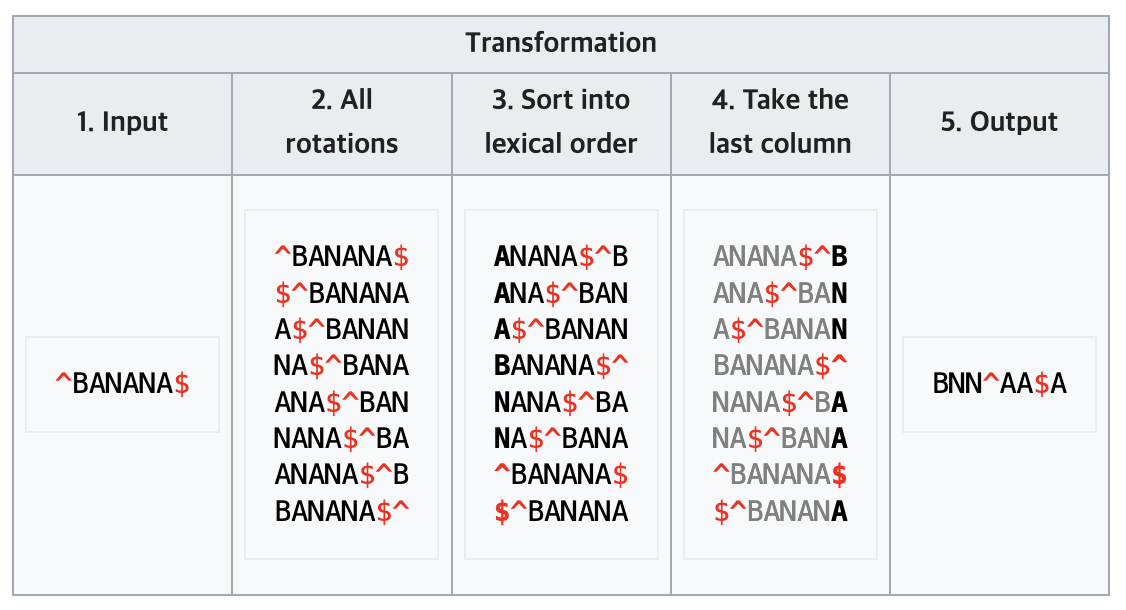
원본 문자열에 대한 모든 Rotation을 만들어 생성된 모든 Rotation 문자열을 알파벳 순서로 정렬 한다.
정렬된 문자열에서 마지막 Column 값들만 취한다.
@ 문자는 ‘EOF’를 의미한다.
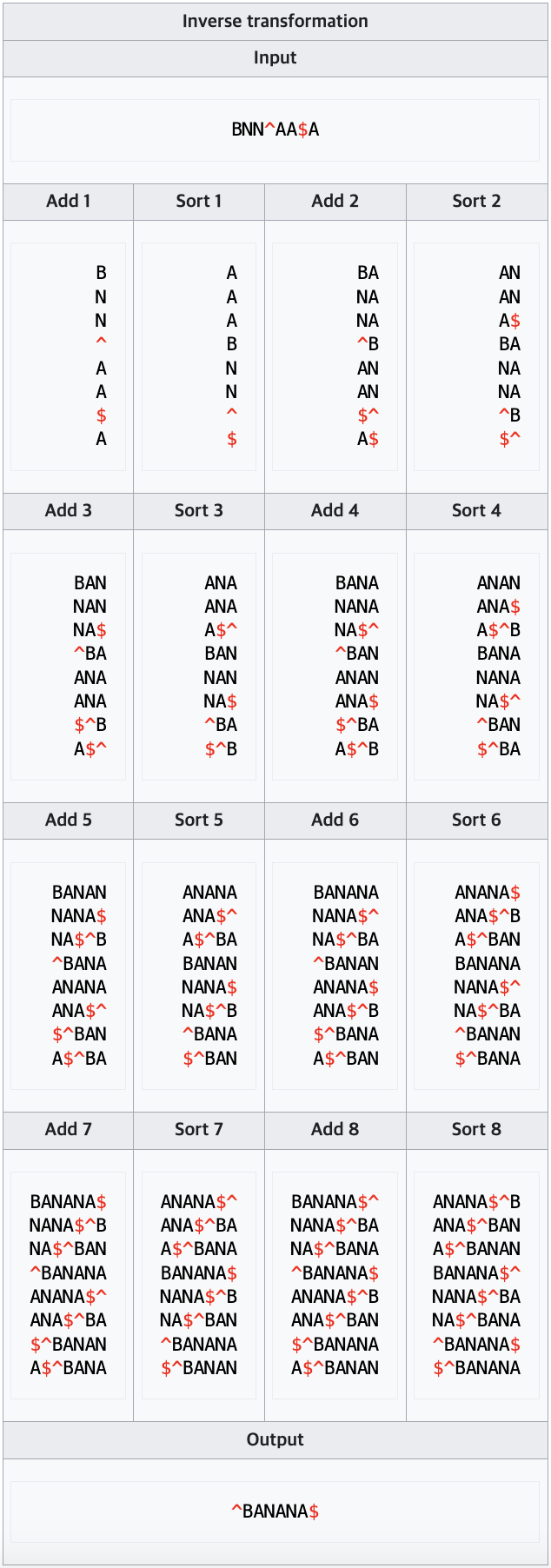
BWT 의 특성은 쉽게 encoded 되는 output 의 생성에 있지 않고, 원본 document 를 재생성 하는 reversible 에 있다.
Encoded 데이터로 부터 원본 문자열을 추출하는 방법은 아래와 같다.
최종적으로 transform 되어진 문자열을 일렬로 나열하고 알파벳 순으로 정렬한다.
다시 transform 된 문자열을 붙이고 정렬작업을 다시 한다.
이와 같은 방식으로 전체 리스트를 구성하여 ‘EOF’ 문자열 ‘@’ 이 가장 마지막에 들어있는 문자열이 원본 문자열이 된다.
2000년대 후반 High-throughput sequencing(HTS) 발전은 BWT의 새로운 응용분야를 만들었다.
HTS 에서 DNA 는 수 백만 개의 reads 조각(30~500 base pairs)으로 fragmented 되어 sequencing된다.
ChIP-Seq과 같은 많은 실험에서 이 reads들을 reference genome에 align 한다.
이러한 task에 관련된 수많은 alignment program들이 publish 되었다
최초에 hashing 방법을 사용하였을 때에는 매우 큰 메모리 용량을 필요로 하기 때문에 alignment-program의 next-generation은 효과적인 reads 의 align을 위하여 reference genome의 burrows-Wheeler transform을 활용한다.
'Bioinformatics' 카테고리의 다른 글
| MapSplice2 Alignment 분석 도구 설치 및 실행 방법 (0) | 2017.01.06 |
|---|---|
| BLAST Nr/Nt 데이터베이스에서 시퀀싱 서열로 변환 명령어 (0) | 2016.02.17 |
| Burrows-Wheeler Aligner(BWA) Alignment 분석 도구 설치 및 실행 방법 (0) | 2012.07.28 |
| SRA Toolkit 을 이용한 SRA 파일 FASTQ 로 전환하기 (0) | 2012.06.07 |
| Bowtie Alignment 분석 도구 설치 및 실행 방법 (0) | 2011.03.14 |
Korean BioInformation Center(KOBIC) Korea Research Institute of Bioscience & Biotechnology Address: #52 Eoeun-dong, Yuseong-gu, Deajeon, 305-806, KOREA +82-10-9936-2261 e-mail: kogun82@kribb.re.kr Blog: kogun82.tistory.com Homepage: www.kobic.re.kr
포스팅이 좋았다면 "좋아요❤️" 또는 "구독👍🏻" 해주세요!



